
Type 2 diabetes is one of obesity’s most tightly linked comorbidities, and its prevalence is rising in parallel with the global obesity epidemic. Uncontrolled obesity often leads to insulin resistance, beta cell failure, and hyperglycemia. Although effective glucose-lowering agents, almost all circumvent rather than address the pathophysiology of this disease. The thiazolidinediones (TZDs) are a class of anti-diabetic drugs that are unique as they are insulin sensitizers capable of delaying or even preventing the onset of diabetes. Despite potent action on insulin sensitization, clinical use of TZDs has declined due to adverse side effects. As ligands for the nuclear receptor peroxisome proliferator activated receptor-γ (PPARγ), the therapeutic actions of TZDs have been assumed to result from their classical agonism towards PPARγ to drive adipocyte differentiation. PPARγ is highly expressed in adipose tissue, where it was first characterized as a master regulator of adipocyte differentiation and function. Activation of PPARγ improves insulin sensitivity owing to diverse roles on both adipocyte and non-adipocyte cells, including macrophages, T-cells, muscle, and others. PPARγ is intimately linked to insulin sensitivity, and genetic variants in the PPARG gene locus are found to associate with diabetes risk.
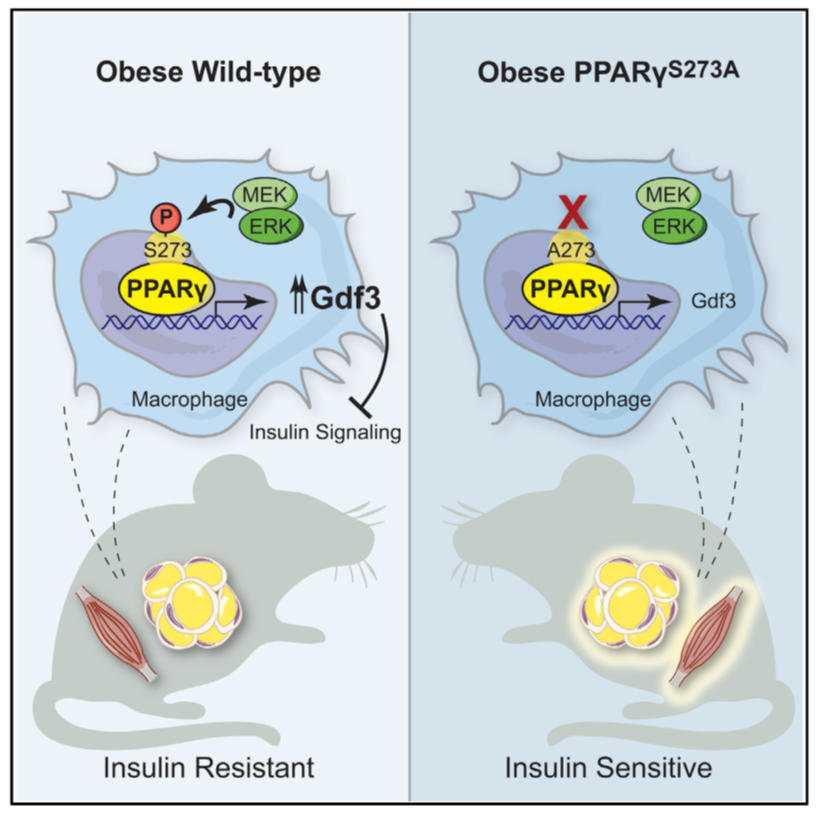
Phosphorylation of PPARγ at serine 273 (p-S273) is observed in adipose tissue shortly after the initiation of high-fat diet feeding and levels continue to increase with progressive obesity. When diabetic patients or mice are treated with TZDs, the phosphorylation of PPARγ S273 is blocked, gene expression profiles are normalized, and insulin sensitivity improves (Nature 2010). Furthermore high-affinity ligands of PPARγ designed to only block p-S273, still retain anti-diabetic effects but lack activity on adipogenesis (Nature 2011). These “non-agonist ligands” are safe and do not trigger the side effects associated with TZDs use. Likewise, in vivo inhibition of the kinase responsible for PPARγ S273 phosphorylation, extracellular signal-regulated kinase (ERK), improves insulin sensitivity in obese animals (Nature 2015). These experiments had suggested a causal role for PPARγ p-S273 to promote insulin resistance. We now firmly establish this causal link with a strain of mice (PPARγ Serine 273 to Alanine global knock-in, PPARγ A/A) which cannot be phosphorylated at S273 and also retain insulin sensitivity (Cell Metabolism 2020). Our model demonstrates that PPARγ p-S273 is sufficient to impart obesity-mediated insulin resistance. Quite unexpectedly, we observe a highly specific downregulation of Growth Differentiation Factor 3 (Gdf3) in epididymal white adipose tissue from PPARγ A/A mice. The significance of this work is that targeting PPARγ phosphorylation can safely and strongly improve insulin sensitivity.